


1 AlphaFold与结构生物学
近年来,人工智能(artificial intelligence,AI)以惊人的速度发展,改变了我们生活和科学研究的许多方面。2024年诺贝尔物理学奖和化学奖双双花落AI领域,物理学奖突出“科学如何应用于AI,改变AI”,而化学奖突出“AI如何改变科学和人们的认知”。本文将探讨获得2024年诺贝尔化学奖的蛋白质结构预测工具AlphaFold和传统的结构生物学方法的异同。
AlphaFold是由DeepMind公司开发的AI模型,能够根据蛋白质的氨基酸序列预测其三维结构。蛋白质就像是生命体内的小机器,它们的结构决定了功能。了解蛋白质的结构对于药物研发以及理解生命过程非常重要。AlphaFold的出现,让人们看到了快速预测蛋白质结构的可能性。
截至目前,AlphaFold的3个主要版本是AlphaFold1、Alpha Fold2和AlphaFold3,各自代表了从基础探索到高精度预测和复合体建模的逐步演进(表 1)。Al phaFold1于2018年推出,采用卷积神经网络处理多重序列比对数据,并结合物理化学约束预测蛋白质三维结构,但在复杂和长链蛋白中精度有限[1]。AlphaFold2在2020年问世,使用Transformer和图神经网络相结合的架构,通过Evoformer模块整合序列信息并直接优化原子坐标,大幅提高了预测精度,在单体蛋白结构预测中接近实验解析水平[2]。Al phaFold3于2024年推出,作为DeepMind公司在蛋白质结构预测领域的最新突破,相较于前2代模型实现了显著的功能扩展和性能提升,其模型架构引入了全新的“Pairformer”模块,进一步提升了AlphaFold2中的Evoformer模块。同时结合扩散模型,从原子点云的初始状态出发,迭代生成分子结构的三维表示。这一创新极大地提高了复杂生物分子建模的效率和精度,且能够预测带配体的复合物结构[3]。
表1 3代AlphaFold的核心差异对比 |
| 版本 | 架构特点 | 关键技术 | 应用和性能 |
| AlphaFold1 | 卷积神经网络 | 序列对齐信息、物理化学约束 | 精度有限,局限于简单蛋白 |
| AlphaFold2 | Transformer+图神经网络 | Evoformer、结构模块、注意力机制 | 精度接近实验水平,适用于广泛单体蛋白预测 |
| AlphaFold3 | Pairformer+扩散模型 | 蛋白质与DNA、RNA、配体、离子等相互作用预测,翻译后修饰影响 | 提高复合物预测精度,助力药物设计和生物学研究 |
传统上,结构生物学使用实验手段来解析蛋白质的三维结构,主要方法有以下几种。
X射线晶体学:这是最早也是最常用的方法。研究人员首先需要让蛋白质形成晶体,这就像把很多相同的蛋白质整齐地排列在一起。然后,用X射线照射这些晶体,得到衍射图样,通过解析这些图样计算出蛋白质的三维结构。这一过程非常复杂,需要大量的时间和精力,尤其是培养出合适的蛋白质晶体并不容易,并且某些蛋白质无法在任何条件下结晶,限制了晶体学对蛋白结构的研究[4]。
核磁共振(nuclear magnetic resonance,NMR):这种方法利用原子核在磁场中的特性。研究人员将蛋白质溶解在溶液后放入强大的磁场中,然后测量原子核的信号。通过这些信号,可以推断出蛋白质的结构和动态信息。NMR适用于研究小型蛋白质,并且可以观察到蛋白质在溶液中的自然状态。但是,对于分子量较大的蛋白复合体,NMR的方法并不适用[5]。
冷冻电子显微镜(cryo-electron microscopy,Cryo-EM):这是近年来迅速发展的技术。研究人员将蛋白质快速冷冻,保持其天然状态,然后在电子显微镜下观察。电子显微镜能提供非常高的分辨率,甚至可以看到单个原子的排列。但总体上精度不如晶体学研究,仅是在部分结构中达到了近原子分辨率。Cryo-EM特别适合研究大型的蛋白质复合物,但设备昂贵,操作也需要高超的技术[6]。
这些传统方法虽然精确可靠,但过程繁琐,耗时耗力,需要丰富的经验和技术支持。因此,当AlphaFold这样的AI工具出现后,一些人开始思考:既然我们可以快速预测蛋白质结构,传统的实验方法是否还有必要呢?
虽然AlphaFold在很多情况下能给出较为准确地预测,但它也有局限性。首先,蛋白质并不是一成不变的,它们会随着环境的变化而改变构象。AlphaFold对于这种动态变化的预测能力有限。其次,许多蛋白质需要与其他分子相互作用才能发挥功能,形成复杂的复合物。预测这些复合物结构,对AlphaFold来说仍然是巨大的挑战。因此,AI的预测结果可能依然需要通过实验来确保其准确性。
作为结构生物学的研究者,徐华强团队深入对比了结构预测工具和实验结构,并发文[7]比较了AlphaFold预测的G蛋白偶联受体(G protein-coupled receptors,GP CR)结构与实验解析的结果。研究发现,虽然AI预测有一定的准确性,但在关键细节上仍然存在差异。例如,某些重要的活性部位,预测结果可能并不准确。这些细微的差别,对于药物设计和功能研究可能会产生重大影响。
AlphaFold的出现是科学发展的重要里程碑,为我们提供了强大的工具。然而,它并不能完全取代传统的结构生物学方法。实验验证和深入研究仍然是理解生命奥秘的关键,在研究中,我们需要拥抱新技术,但也要意识到其局限性。
2 GPCR与AlphaFold2
GPCR是一种通过G蛋白传导信号的受体,广泛表达于细胞膜表面,负责将胞外信号传递到细胞内部。人类能看到东西、闻到味道,甚至感受到情绪波动,如开心和难过,GPCR都在其中扮演着关键角色。正因如此,它成为了现代药物设计中最重要的靶点之一,美国食品药品监督管理局(Food and Drug Administration,FDA)批准的药物中约有1/3都作用于GPCR,其年销售额超过1万亿美元[8]。
在这一背景下,DeepMind公司开发的AlphaFold2为GPCR结构预测带来了革命性的突破。AlphaFold2在蛋白质结构预测竞赛中表现出色,展示了接近实验精度的预测效果。AlphaFold2的成功不仅证明了AI在生物学研究中的巨大潜力,也为GPCR相关的药物设计和功能研究提供了强有力的工具。
然而,尽管AlphaFold2在结构预测方面取得了一定成就,它在取代传统结构生物学方法上仍面临不少局限。本文选取了Al phaFold2发表后的29个GPCR结构,使用AlphaFold2折叠了它们的预测模型,并与实验结构进行比较和评测。由于这些蛋白不在训练集中,这排除了AlphaFold2预测时参考训练集数据的可能。
3 整体结构区别:Alpha Fold2是个好导航,但可以更好
在细胞生物学的世界里,蛋白质像一台复杂的机器,每个零件都至关重要。GPCR作为细胞表面的受体,更像是传递外界信号的特工。AlphaFold2则是高科技的导航系统,能够预测这些特工的“路线”,帮助科学家理解它们的工作方式。但是,就像导航软件提供的路线与实际道路可能存在差异一样,我们仍然需要结合实际的情况来作出正确的判断。
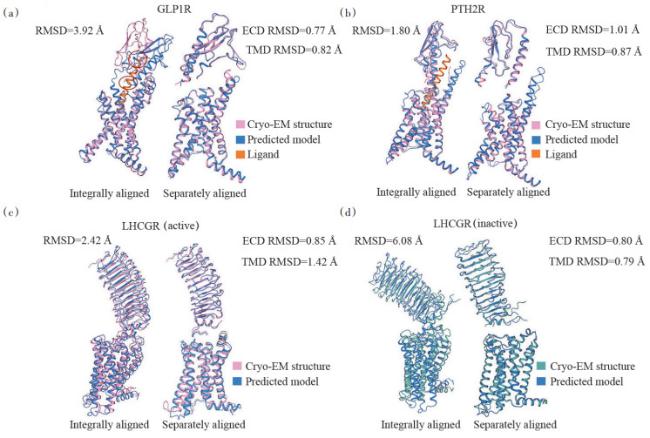
GPCR由7段跨膜螺旋组成,就像一条包含7个关键路段的复杂路线。这些螺旋在细胞膜中各自“行驶”。螺旋1~4像在主干道上稳定前进的车辆,而螺旋5~7则像是在交通状况变化时出现的绕行路线,随着激活状态的变化而表现出更多的“动态”。Alpha Fold2在捕捉这条“路线”的整体布局上表现得相当不错。我们评测的这些蛋白整体均方根偏差(root mean square deviation,RMSD)仅为1.64 Å,显示出Al phaFold2在解读GPCR复杂结构上的精确度。
然而,真正的挑战并不在于稳定的7次跨膜区域。当这些GPCR带上巨大的“附加装置”——大型的细胞外结构(extracel- lular domain,ECD)时,AlphaFold2的预测就像是试图将一辆装满货物的卡车通过一个狭窄的隧道。这些ECD结构的预测误差通常会增大,因为ECD和跨膜区域(transmembrane domain,TMD)之间的相对位置预测不够准确,就像货车与隧道未能正确对齐一样。例如,结合了semaglutide的胰高血糖素样肽-1受体(glucagon-like peptide-1 receptor,GLP1R),其整体误差达到了惊人的3.92 Å。在甲状旁腺激素2受体(parathyroid hormone 2 recep tor,PTH2R)和激活态的黄体生成素/绒毛膜促性腺激素受体(luteinizing hormone/choriogonadotro pin receptor,LHCGR)中,也出现了整体RMSD大于分开计算的RMSD的问题。对于在训练集中不常见的失活态LHCGR,整体RMSD竟然达到了6.08 Å,超过了2根半氢键的长度,这表明二者的差异极大,类似于按照AlphaFold的导航在高速公路上误入了逆行车道(图 2)[7]。
4 关键药物作用位点:AlphaFold2无法指引重要路口的方向
GPCR就像一座繁忙的城市中心,而小分子药物就是来自各地的游客,试图找到这座城市中最关键的交汇点——正构位点。在这里,小分子与GPCR的互动就像是在重要的路口触发了交通信号,进而改变了细胞内的“交通流向”,对细胞功能产生重大影响。因此,准确了解这个关键“路口”的结构,对于基于结构的药物设计和功能研究至关重要。
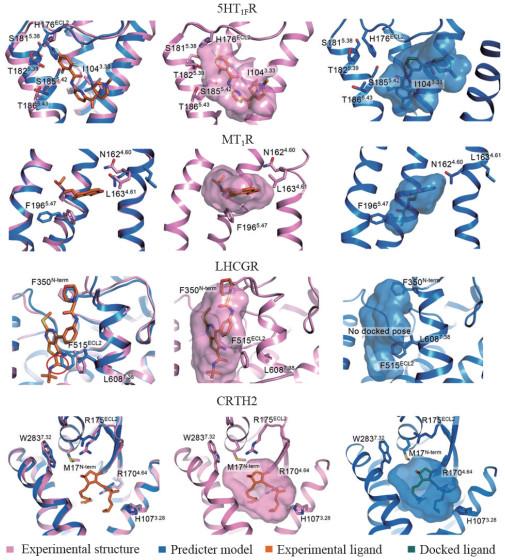
在5-羟色胺1F受体(5-hy droxytryptamine receptor 1F,5HT1FR)的案例中,AlphaFold2预测的侧链排列出现了偏差,就像导航错误地引导车辆进入了狭窄的街道。一些侧链朝向细胞膜的中心“转向”,使得原本宽敞的“道路”变得拥挤不堪。这种变化阻碍了三氟苯环与H176ECL2的相互作用,导致分子对接生成的小分子结合姿态与实验结构大相径庭,RMSD达到了7.15 Å,也完全没有重现相互作用。
在褪黑素受体1A(melatonin receptor 1A,MT1R)的案例中,F1965.47的侧链向外“偏航”,形成了一个意外的“隧道”,使得小分子更深入地“驶入”了受体内部。此外,其他几个残基的侧链也发生了调整,导致对接后的小分子朝着TM螺旋束的中心移动,最终RMSD为4.79 Å。这就好比导航引导我们进入了一条未知的地下通道,偏离了预期的路线。
在LHCGR的案例中,尽管主链结构差异很小,侧链的不同却成为了关键的“路障”。F515ECL2的侧链插入了TM5和TM7之间的“通道”,改变了顶部“交叉口”的环境,导致小分子配体甚至无法在预测模型中成功被对接。如果基于这些结构进行药物设计,结果可能就像是在堵塞的道路上行驶,无法到达目的地。
当然,在2型辅助T细胞上表达的趋化受体同源分子(chemoat-tractant receptor-homologous molecule expressed on T-Helper type 2 cells,CRTH2)的案例中,预测模型与实验结构在正构位点的主链和侧链都高度一致,对接结果也显示小分子的预测结合姿态与实验数据几乎完全吻合,RMSD仅0.90 Å。然而,这种理想情况并不能在所有预测模型中得到保证。
5 TM6-TM7:复杂多变的导航路线,AlphaFold2往往难以把握
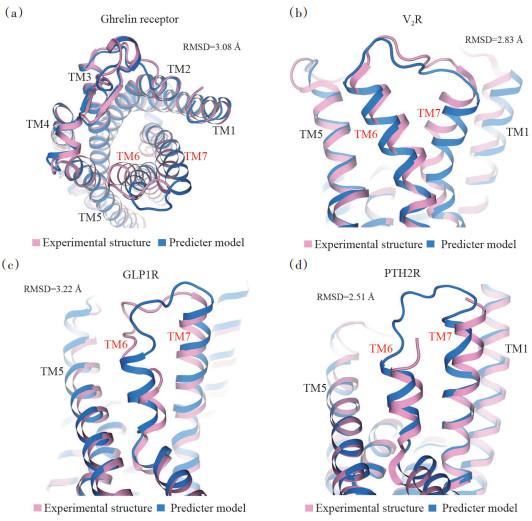
首先,研究这些“交通枢纽”在细胞外侧区域的差异发现,有些GPCR在预测模型中的TM6-TM7构象与实验结果有较大出入,误差超过2 Å。例如,在ghre lin受体和抗利尿激素受体(vaso pressin receptor 2,V2R)的“地图”中,这些关键“路段”的偏差分别达到了3.08 Å和2.83 Å,仿佛导航系统给出了与实际道路不符的指引。
在GLP1R和PTH2R的模型中,TM6和TM7被预测为“向上抬升”,与实验结构形成鲜明对比。这种“向上”的变化就像导航错误地显示了一座不存在的高架桥,导致原本应该“通行”的小分子无法正确“到达”结合位点,这对药物设计来说是个重大障碍。

相比之下,B1类GPCR的预测模型与实验结构几乎完全一致,说明AlphaFold2在这部分的“地图绘制”非常准确。这可能是因为训练数据中包含了较多的激活态B类GPCR结构,就像导航系统在这些区域的数据更加丰富且可靠。
另外需要注意的是,某些A类GPCR的胞内环区3(intracellu lar loop 3,ICL3)在预测模型中被“描绘”成了一段长长的“直路”,这与实验结构中的“蜿蜒小路”大相径庭。例如,5HT1FR和胆囊收缩素受体1(cholecystokinin A receptor,CCKAR)就出现了这种情况,就好比导航错误地告诉我们前方是一条笔直的大道,实际上却是一段曲折的山路。
6 蛋白结构预测的误差:为什么AlphaFold2有时会“迷路”?
在AlphaFold2的视角中,GP CR蛋白就像一张复杂多变的地图,但有时它会在某些关键区域给出与实际情况不符的“导航指引”。这些预测模型中不同的ECD和TMD的组合,就好比导航系统生成了与真实道路不一致的路线,虽然有时与驾驶者(科学家们)的预期不符,但也可能揭示了一些尚未被发现的、短暂存在的“道路”状态。
例如,在GLP1R的案例中,预测的ECD-TMD结构阻碍了肽的结合。这种误差可能是由于Al phaFold2在训练过程中缺乏足够的配体信息,导致无法准确重现有利于肽结合的特定ECD-TMD构象,就像导航系统的地图数据不完整,无法提供最佳路线。
预测与小分子结合的GPCR结构时,尽管主链的预测准确度达到1 Å左右,但这和GPCR本身的保守性关系很大。AlphaFold2在预测与配体相互作用的“结合口袋”结构时仍面临挑战。更糟糕的是,在LHCGR的案例中,预测模型甚至未能形成适合小分子对接的“停靠点”,就如同导航缺少了关键的目的地信息,让的旅程无法完成。如果基于这样的“地图”去设计药物,无异于在错误的地点寻找目标。
对于TM6螺旋的预测,Al phaFold2似乎倾向于产生一种介于激活态和非激活态之间的“平均”构象。这种数据偏差导致的预测结果,就像导航系统给出了2条路线的折中方案,但其实只有2条道路可以走,中间这条路是不稳定,走不通的。此外,ICL3区域的预测也常常出现过长的螺旋结构,而在实验结构中,这些区域通常是灵活多变的。这可能是因为AlphaFold2从包含骨限制性干扰素诱导跨膜蛋白样(bone-restricted interferon induced transmem brane protein-like,BRIL)融合蛋白的非天然GPCR结构中学习。
7 未来研究,要谨慎使用AI模型
通过这些例子,我们认识了AlphaFold2在GPCR结构预测中的局限性,作为从提出到获得诺贝尔奖的最快例子之一,Alpha Fold2为研究领域带来了革命性的变化,但仍不能忽视其潜在的问题。在未来的研究中,科学家们需要谨慎地使用这些预测模型,结合实验结构生物学的方法,进行配体结合位点和激活机制的相关验证,以为真实的药物设计提供参考。AlphaFold2为我们提供了探索蛋白质结构奥秘的工具,但同时也提醒我们,在拥抱新技术的同时,仍需脚踏实地,通过实验发现真实蛋白构象,共同绘制出更精确的蛋白质“路线图”。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}