


1 AI驱动蛋白质结构预测:AlphaFold3的突破与应用
蛋白质是生命活动的功能执行者。精确的蛋白质结构是解析疾病机制、设计靶向药物的基础。传统的蛋白质结构解析方法虽然可以获得高分辨率的蛋白质结构,但存在耗时耗力、通量低等局限性。因此,长期以来,研发人员迫切需求准确高效的蛋白质结构预测方法。AlphaFold3等AI模型的出现,提高了蛋白质结构预测的精度和速度,被认为是加速药物发现的关键技术[6]。
1.1 AI在蛋白质结构预测中的应用
DeepMind公司开发的AlphaFold系列模型是AI驱动蛋白质结构预测领域的里程碑式突破。AlphaFold2的预测精度可以与实验方法相媲美。2024年,DeepMind发布了AlphaFold3,进一步拓展了结构预测的能力[7]。
AlphaFold3不仅能够预测蛋白质的三维结构,还能预测包括脱氧核糖核酸(DNA)、核糖核酸(RNA)、配体以及修饰在内的生物分子复合物的结构。其采用了更先进的Pairformer注意力机制,能够基于成对表示处理高效地捕捉蛋白质序列中的长程依赖关系,并利用更全面的生物学数据库进行训练[8]。
AlphaFold3在多个评估指标上超越AlphaFold2,尤其是在预测蛋白质−配体等复合物结构方面取得了0到1的突破。这为研究复杂的生物分子相互作用,及开发靶向特定复合物的药物提供了强有力的工具[7]。
除了AlphaFold系列模型,蛋白质结构预测领域还涌现出多种高性能模型,包括RoseTTAFold[9]和Protenix等。RoseTTAFold是David Baker实验室推出的深度学习蛋白质结构预测框架,核心创新在于同时更新序列、配对信息及三维坐标的“三轨”并行注意力网络。Protenix是由字节跳动公司开发的AlphaFold3开源实现版本,旨在提高模型的可训练性和可扩展性。该模型在预测蛋白质−配体和蛋白质−核酸相互作用方面表现出色,且开源了训练代码,便于复现。
1.2 结构预测工具在药物研发中的最新研究成果
结构预测工具的突破性进展为药物研发带来了前所未有的机遇。本文以AlphaFold3为例,分析AI在药物研发中的最新研究成果。
首先,在靶点发现与验证方面,AlphaFold3能够帮助研究人员快速获取与疾病相关的蛋白质结构模型。有了这些预测结构,研究人员可以初步理解蛋白质的功能机制,识别潜在的药物结合位点,从而加速靶点发现和验证的过程,然而这一过程通常还需要结构生物学的方法进一步验证[10]。
在虚拟筛选与先导化合物发现方面,AlphaFold3同样展现了巨大价值。传统的虚拟筛选通常依赖已知的蛋白质结构,现在,研究人员可以利用AlphaFold3预测的结构来进行筛选。通过结合分子对接等方法,可以筛选出潜在的先导化合物,提升发现效率。
抗体药物设计是另一个受益匪浅的领域。AlphaFold3能预测抗体−抗原复合物结构,让研究人员得以深入理解抗体与抗原的结合机制,并据此初步指导抗体的序列优化、亲和力成熟以及人源化改造。
此外,AlphaFold3在酶工程与蛋白质药物设计中也可以应用。它不仅能预测天然蛋白质的结构,还能用于分析工程化改造后的蛋白质结构。这为研究人员提供了强有力的工具,来指导酶的定向进化或新型蛋白质药物的设计[11]。
1.3 AlphaFold3的局限性与挑战
2 AI驱动蛋白质设计:从结构预测到功能创新
2.1 AI在蛋白质设计中的应用
AI在蛋白质设计中的核心优势在于能够结合序列生成、结构预测和功能优化,显著提升效率和成功率,图 2描述AI在蛋白质设计中的常见应用场景。
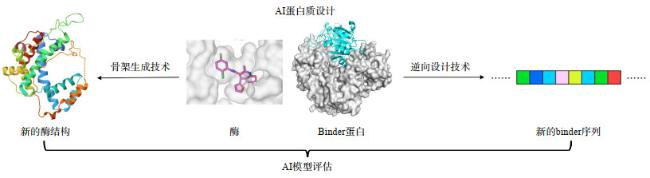
首先,在序列生成与优化方面,AI通过逆向设计技术,可以从目标结构出发生成相应序列,使用生成模型也可以快速设计具有目标功能的蛋白质序列[14]。
其次,在结构导向的设计方面,AI利用蛋白质结构预测的突破进一步推动了设计能力。例如,基于深度学习模型ProteinMPNN能够学习蛋白质序列与功能之间的映射关系,生成具有特定催化活性或结合能力的序列。这些序列随后可通过结构预测工具进行折叠验证,确保其三维结构与预期功能相匹配[15]。
在功能预测与验证方面,AI模型能够评估蛋白质的潜在功能,或优化已有蛋白质的功能。例如,AI可以预测酶的催化效率、蛋白质的结合亲和力或热稳定性,从而筛选出最优候选分子。这种能力在酶工程和治疗性蛋白质开发中尤为重要[16]。
2.2 AI驱动蛋白质设计的最新研究成果
近年来,AI在蛋白质设计领域取得了显著进展。David Baker团队开发的RFdiffusion模型已成功设计出多种新型蛋白质,包括高效催化酶和具有特定结合能力的蛋白质分子[17]。
在工业应用方面,AI驱动的蛋白质设计平台正逐步成熟。例如,Ginkgo Bioworks等公司开发了基于AI的酶设计工具Ginkgo Enzyme Services,通过优化结构显著提升了工业酶的催化效率。
2.3 AI驱动蛋白质设计面临的挑战
尽管AI在蛋白质设计领域展现出巨大潜力,但其发展仍面临若干关键挑战。
首先,数据依赖性是主要瓶颈。AI模型的性能高度依赖于高质量的训练数据。然而,当前的实验数据在多样性上依然不足。训练数据往往局限于天然蛋白质的已知结构和功能。数据稀缺可能导致模型在面对全新设计任务时的泛化能力不足,限制了其在创新性蛋白质设计中的表现。
其次,功能预测的准确性仍需提升。虽然AI工具如AlphaFold3在蛋白质结构预测上取得了突破,但在预测蛋白质功能方面仍存在局限。蛋白质功能的实现往往涉及复杂的动态过程和环境依赖性,而当前AI模型主要基于静态结构预测,难以准确捕捉这些动态特性。这种预测误差可能导致候选分子在实验验证中失败,增加研发成本。
此外,实验成本的高昂是另一大制约因素。AI驱动的蛋白质设计通常需要生成数千个以上候选序列,设计的候选分子需要通过湿实验验证,而这些实验步骤不仅耗时耗力,还涉及昂贵的试剂和设备成本。这使AI驱动蛋白质设计的广泛应用受到一定限制。
3 AI赋能抗体药物设计:加速研发进程,提升药物疗效
抗体药物在生物制药领域中占据重要地位。其凭借靶向性强、特异性高等优势,在肿瘤、自身免疫疾病和感染性疾病等领域的治疗中展现了显著效果[18]。但传统的抗体药物研发依赖于杂交瘤技术、噬菌体展示技术等,面临着周期长、成本高、成功率低的难题。AI技术在抗体序列优化、人源化改造、亲和力成熟和新型抗体设计等方面的应用,有望加速抗体药物的研发进程,提高抗体药物的疗效和安全性。
3.1 AI在抗体药物设计中的应用
AI技术在抗体药物设计的各个环节都展现出强大潜力。首先,在抗体序列优化方面,AI能够提升抗体疗效并降低免疫原性。例如,深度学习模型能够分析抗体互补决定区(complementarity−determining region,CDR)序列与抗原表位的相互作用,优化CDR设计以增强抗体结合能力。
抗体亲和力成熟是另一个AI发挥作用的领域。传统实验方法效率较低,且难以穷尽所有突变组合。而AI技术可以通过机器学习模型,根据抗体序列和亲和力数据预测最佳突变位点和组合,指导定向进化实验,从而提升亲和力成熟的效率和成功率[19]。
在新型抗体设计方面,生成对抗网络通过生成器和判别器的对抗训练学习真实数据分布,生成器可从随机噪声中采样并输出新的抗体序列,从而实现抗体设计的多样性。该技术可以生成全新的抗体序列,并结合预测模型筛选出最具潜力的候选分子[20]。
近年来兴起的扩散模型(diffusion models)通过逐步去噪生成蛋白质三维结构,已被用于精确设计抗体骨架和优化CDR区结构,从而提高抗体与抗原结合的空间匹配度。这类模型不仅能生成符合物理化学约束的新结构,还能在结构空间中探索新颖结构,从而拓展抗体设计空间。
图神经网络(graph neural networks, GNN)能够将抗体和抗原表示为图,通过学习残基之间的空间与化学相互作用,实现亲和力预测与界面识别。例如,GearBind是一种可预训练的几何图神经网络[21],专为抗体亲和力成熟设计,显著提升了对抗体−抗原相互作用的建模能力。
语言模型(language models)也已成为抗体序列设计的重要工具。例如,Hie等[22]使用蛋白语言模型,无需结构或抗原信息即可高效指导抗体亲和力成熟,在仅2轮实验中将抗体亲和力提升最高达160倍。抗体可开发性预测也是AI的重要应用方向。抗体药物的表达量、稳定性、聚集倾向等特性直接影响研发成功率。AI模型通过学习序列与可开发性间的关系,能在早期阶段预测这些特性,排除问题分子。
为便于系统比较不同AI模型在抗体药物设计中的应用特征、技术机制与代表性成果,表 1总结了扩散模型、图神经网络、生成模型与语言模型的核心原理、主要应用环节、优势。
表1 AI模型在抗体药物设计中的应用对比 |
| 模型类型 | 技术原理简述 | 主要应用环节 | 优势 |
| 扩散模型 | 通过逐步添加/去除噪声学习分布,实现蛋白三维结构的高精度生成 | 抗体骨架设计、CDR结构建模 | 可精确建模抗原表位几何约束,探索新结构构象 |
| 图神经网络 | 将分子结构建模为图,学习节点(残基)和边(相互作用)之间的拓扑关系 | 抗体−抗原互作预测、亲和力预测 | 能处理抗体−抗原结构复杂性,支持界面残基识别与突变筛选 |
| 生成对抗网络 | 编码抗体序列到潜在空间或训练对抗网络,生成结构合理的新抗体序列 | 抗体序列生成、亲和力多样性探索 | 可合成高质量且多样的抗体序列,辅助构建抗体库 |
| 语言模型 | 使用蛋白序列作为输入,基于自注意力机制学习序列和功能信息 | 序列优化、指导亲和力成熟 | 适用于大规模数据、快速评估突变影响 |
3.2 AI驱动抗体药物设计的最新研究成果
近年来,AI在抗体药物设计领域的进展令人瞩目。例如Absci、Adimab和Ginkgo Bioworks等公司开发的平台,利用深度学习优化抗体,并在实验中得到验证,极大提升了开发效率。其中,Mason等[23]开发的一种端到端序列优化方法,通过深度神经网络模型在108规模的虚拟抗体库中筛选抗原特异性抗体,随机抽取的30个突变体中全部表现出对HER2的有效结合,显示出AI在高通量抗体筛选中的强大能力。
在抗体可开发性预测方面,AI工具同样取得了突破。Liang等[26]开发的IsAb2.0平台结合深度学习与多目标序列优化算法,成功实现了对纳米抗体J3的人源化重设计。通过引入少量优化突变,获得了热稳定性更高、表达水平更优且保留功能活性的HuJ3抗体变体,证明了AI在人源化路径规划中的实用性。
3.3 AI驱动抗体药物设计面临的挑战
尽管AI在抗体药物设计中表现亮眼,但仍面临一些难题。数据质量和数据量是首要挑战。AI模型依赖大量高质量数据,虽然抗体序列的数据量较大,但其亲和力数据相对稀缺,且质量不一。未来需要建立更大规模、更优质的抗体数据库,同时开发数据增强和挖掘技术。
4 AI助力小分子药物设计:加速发现、优化性质、提高效率
小分子药物是药物研发领域的核心力量,占据了药物市场的绝大部分份额。传统的小分子药物研发的方式效率较低,研发周期长。AI技术在小分子药物的靶点识别、虚拟筛选、先导化合物优化,以及药物的吸收(absorption)、分布(distribution)、代谢(metabolism)、排泄(excretion)和毒性(toxicity)——即ADMET性质预测等方面,正发挥着日益重要的作用。AI的应用有望提升研发效率,并降低研发成本[4]。
4.1 AI在小分子药物设计中的应用
AI在小分子药物设计的每个环节都展现出一定潜力。在靶点识别与验证方面,AI能够分析海量生物学数据,包括基因组、转录组、蛋白质组和代谢组数据,找出已知产生药效的小分子的潜在靶点,并评估其可药性[29]。
先导化合物优化同样受益于AI。先导化合物往往存在活性不足、选择性差或ADMET性质不佳等问题。AI模型能够预测这些特性,指导结构修饰,提升成药性[32]。
小分子药物的合成路线设计也因AI而更高效。AI可以预测反应产率和选择性,优化路线。例如,基于强化学习的模型可以自动探索合成路径,找到成本最低、效率最高的方案[33]。
4.2 AI驱动小分子药物设计的最新研究成果
近年,AI在小分子药物设计领域成果斐然。基于深度学习虚拟筛选平台,如Atomwise和Exscientia可以预测化合物活性,已在多个项目中加速了先导化合物发现。例如,Exscientia与Sumitomo Dainippon Pharma公司合作开发的DSP−1181用于强迫症治疗,已进入临床试验,仅用12个月,远快于传统的5年时间。
AI驱动的先导化合物优化技术也不断突破。基于生成模型和强化学习的方法能够高效探索化合物结构空间,显著提升成功率。例如,Insilico Medicine公司的GENTRL模型利用生成性张量强化学习在21 d内设计出有效的DDR1激酶抑制剂,总耗时仅46 d,远超传统方法[34]。
AI驱动的药物性质预测工具同样快速发展。基于机器学习的ADMET预测模型和深度学习的毒性预测模型,帮助研究人员在早期评估药物潜力,提高成药性。
4.3 AI驱动小分子药物设计面临的挑战
尽管进展显著,AI在小分子药物设计中仍面临挑战。数据质量和偏差是首要问题。小分子药物的活性与性质数据常存在分布不均、标注错误等问题,影响模型性能。未来需建立更高质量的数据库,并改进数据清洗与增强方法[35]。
模型的泛化能力和鲁棒性也有待提升。AI模型在训练数据上表现良好,但在新数据集上可能失灵,数据集的偏差可能导致模型高性能表现实际上反映了数据偏差,而非物理原理的成功泛化。针对不同靶点和疾病,需开发更具通用性和稳定性的模型。
模型可解释性不足限制了应用。AI预测结果常难以解释,而在药物设计中,理解化学和生物学机制至关重要。开发可解释性更强的模型并提供决策支持工具将是关键。Jiménez−Luna等[36]指出,深度学习模型的“黑箱”性质限制了其在药物发现中的应用,亟需开发可解释性更强的模型并提供决策支持工具。
化学空间探索的局限性也不容忽视。AI基于已知数据生成化合物,可能局限于现有化学空间。未来需结合化学合成和高通量筛选,尤其在竞争激烈的领域如激酶抑制剂发现中探索更广阔的未知领域[37]。
5 AI驱动药物研发的未来展望
未来,AI将在药物研发中扮演更加核心的角色,改变传统研发范式,加速新药发现,降低成本,提升成功率,最终造福人类健康,表 2总结了目前AI药物研发的挑战和进展。
表2 AI驱动药物研发进展与挑战总结 |
| 领域 | 关键进展 | 主要应用场景 | 现存挑战 | 未来发展方向 |
| 蛋白质结构预测 | AlphaFold3实现生物分子复合物结构预测 预测精度接近实验方法 突破性扩展至配体,核酸相互作用预测 | 靶点发现与验证 虚拟筛选与分子对接 抗体−抗原复合物解析 酶工程与药物重定位 | 动态行为预测能力不足 计算资源需求高 实验验证成本高 | 动态构象与相互作用预测 算法优化降低算力需求 多模态数据整合与平台开发 |
| 蛋白质设计 | RFdiffusion实现功能导向的从头设计 生成模型辅助结构改造 工业酶催化效率提升 | 新型治疗性蛋白质开发 酶从头设计 蛋白稳定性优化 合成生物学元件设计 | 非天然蛋白数据稀缺 功能预测准确性不足 实验验证成本高昂 | 多模态数据融合 动态行为预测模型开发 自动化设计−验证闭环平台 |
| 抗体药物设计 | RFantibody实现结构导向 抗体生成 AI亲和力优化 AI辅助可开发性预测 | 抗体人源化优化 亲和力成熟加速 双特异性抗体设计 稳定性与聚集倾向预测 | 免疫原性预测模型不足 动态表位预测困难 实验通量限制验证效率 | 多组学数据整合 可解释AI指导工程化改造 自动化高通量验证平台开发 |
| 小分子药物设计 | AI虚拟筛选与分子生成工具 AI化合物优化平台 ADMET预测模型 | 靶点识别与验证 全新分子骨架生成 合成路线智能规划 毒性预测 | 化学空间探索局限性 活性−选择性平衡困难 多靶点协同作用预测不足 | 知识图谱增强型生成模型 动态药效团建技术 自动化合成−测试闭环系统 |
5.1 蛋白质结构预测工具的未来发展方向
蛋白质结构预测工具有以下方面可以进一步推进。首先是提高预测精度,可以结合生物学领域内的知识与大语言模型,进一步提升其对复杂蛋白质和生物分子复合物的预测能力[8]。
降低计算资源需求并提升计算效率也是重要方向。通过优化算法和模型,未来的AlphaFold3有望在更普通的计算设备上运行,从而惠及更多研究者。
开发用户友好的工具和平台是另一个趋势。可以推出在线的结构预测平台、可视化工具以及与其他药物研发软件的集成接口,让非专业用户也能轻松上手,推动技术普及。
5.2 AI驱动蛋白质设计的未来发展方向
AI驱动蛋白质设计将在以下关键发展方向产生突破,进一步赋能药物研发和生物技术创新。
首先,多模态数据整合是提升设计能力的重要途径。未来的AI模型需要超越单一的序列或结构数据,整合动态变化和细胞层面的多维度信息,以更全面地理解蛋白质的行为。
其次,动态行为预测将成为研究的重点方向。蛋白质的功能往往依赖于其在细胞环境中的动态构象变化。未来的AI工具可能通过结合分子动力学模拟或构象生成模型,预测蛋白质的实时相互作用。
此外,自动化设计平台的开发将显著推动技术普及应用。通过构建集成的全流程平台,AI可形成“设计—表达—测试—优化”的闭环系统,高效完成任务。
最后,与抗体药物设计的协同创新是未来发展的重要趋势。例如,AI可以通过设计新型蛋白框架,为抗体药物提供更多样化的结构选择,蛋白质设计还可用于优化抗体−抗原相互作用界面,通过调整局部结构增强结合亲和力。
5.3 AI驱动抗体药物设计的未来发展方向
5.4 AI驱动小分子药物设计的未来发展方向
未来,AI在小分子药物设计中的发展将更加多元化。通过整合化合物结构、性质、活性及靶点疾病信息,研究者将构建更全面的网络药理学模型,从而提升设计精度[38]。
发展基于知识的AI模型也是可行的方向。将化学和生物学知识融入模型,如规则系统或知识图谱,能提高可解释性和决策支持能力[39]。
AI驱动的自动化药物合成平台将进一步加速研发。通过集成路线设计、反应优化和自动化合成,整个流程将实现智能化,大幅缩短周期。例如,IBM公司的RXN for Chemistry平台已经在利用AI预测化学反应和合成路线,而Chemputer项目已经开发了通用化学机器人来自动化合成过程[40]。
个性化小分子药物设计是未来的亮点。结合基因组学、蛋白质组学和代谢组学数据,AI也有可能为不同患者定制药物,提供更精准的治疗方案[41]。
5.5 AI时代下的药物研发展望
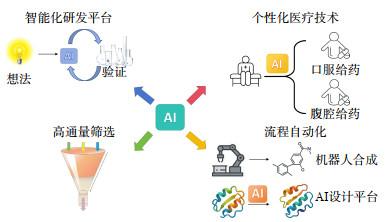
在AI辅助中,药物研发将更依赖多学科交叉融合,AI技术将与生物学、化学、药学、医学和工程学深度结合,构建智能化研发平台。平台不仅能整合基因组、临床、影像等多源数据,通过AI进行分析、建模和优化,有望结合自动化实验技术实现高通量验证,推动从靶点发现到药物优化的全流程高效化。
随着个性化医疗技术的进步,AI将助力精准药物研发,通过分析患者个体化的基因组和临床数据,预测疾病风险和药物反应,设计个性化治疗方案,实现更精准的治疗效果(图 3)。此外,自动化和智能化将成为药物研发的主流趋势,高通量筛选、机器人化学合成与AI设计平台的协同工作,将使研发全流程实现自动化,大幅缩短研发周期,让药物研发最终进入一个全新阶段。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}